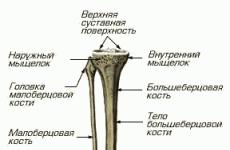
Норильская межрайонная детская больница. Федеральные клинические рекомендации по оказанию помощи детям с гемолитико-уремическим синдромом Жалобы и анамнез
Гемолитико-уремический синдром – клинико-гематологический симптомокомплекс, отличающийся разнообразием этиологических факторов и возникновением тяжелейших нарушений в организме человека. Эта патология характеризуется гемолизом эритроцитов, снижением количества тромбоцитов в крови, дисфункцией почек . Тромботическая микроангиопатия сопровождается множественным образованием тромбов и окклюзией мелких кровеносных сосудов. При этом развивается тромбоцитопения, появляются очаги ишемии и некроза в мозговой ткани и внутренних органах.
Гемолитико-уремический синдром имеет еще одно наименование - болезнь Гассера, полученное в честь своего первооткрывателя С. Gasser в середине прошлого столетия. У пациентов с данной патологией появляется боль в животе, понос с кровью, бледность кожи, иктеричность склер, отечность лица, петехиальная сыпь, анурия, симптомы поражения ЦНС, печени, сердца. Диагностика заболевания основывается на клинических симптомах и результатах лабораторных исследований. Своевременная и грамотная терапия ГУС позволяет сделать прогноз патологии благоприятным.

Болезнь Гассера развивается у грудничков, детей дошкольного возраста, школьников, подростков и очень редко у взрослых людей. У женщин после родов патология протекает наиболее тяжело и часто приобретает рецидивирующий характер. Гемолитико-уремический синдром не имеет определенной сезонности. Заболеваемость достигает максимального уровня в летне-осенний период: с июня по сентябрь. Патология кишечной этиологии регистрируется обычно в летнее время, а заболевания вирусного происхождения – осенью и зимой.
Атипичный гемолитико-уремический синдром (аГУС) - редко встречающаяся патология, которая отличается тяжелым течением и имеет неблагоприятный прогноз. Поражение капилляров, артериол и венул нарушает функционирование внутренних органов, что проявляется разнообразной клинической симптоматикой.
Классификация
Гемолитико-уремический синдром по этиологии и клинике подразделяется на два вида:
- Типичный, взаимосвязанный с диарейным синдромом - Д+,
- Атипичный, не связанный с диареей - Д-.

Диарейные формы возникают у детей младше 5 лет, проживающих в эндемических регионах – Поволжье, Подмосковье. Атипичный ГУС развивается у детей школьного возраста и взрослых лиц.
ГУС может протекать в легкой или тяжелой форме:
- Легкая форма бывает двух типов. Тип А проявляется классической триадой: анемией, тромбоцитопенией и патологией почек. Тип Б проявляется той же триадой, а также судорогами и гипертонией.
- Тяжелая форма подразделяется на два типа. Тип А проявляется триадой симптомов и длится более суток. Тип Б имеет аналогичные симптомы, к которым присоединяются судороги, анурия и выраженная гипертензия.
Этиологическая классификация атипичного ГУС:
- поствакцинальный,
- постинфекционный,
- наследственный,
- лекарственный,
- идиопатический.
Этиология
Типичный ГУС

патогенез бактериальной диареи
Возбудителями ГУС, связанного с диареей, являются энтерогеморрагическая кишечная палочка, шигелла, стафилококк, а более редких случаях - сальмонеллы, кампилобактерии, клостридии.
Эшерихия продуцирует шига-подобный веротоксин, который поражает клетки сосудов почек у детей младше 3 лет. Эндотелиоциты погибают, возникает воспаление, гемолиз эритроцитов, адгезия и агрегация тромбоцитов, развивается ДВС-синдром. Микроциркуляторные расстройства приводят к гипоксии внутренних органов. У больных на фоне ОКИ происходит воспаление клубочков почек, нарушение фильтрационной функции, развитие ишемии, образование очагов некроза, снижение работоспособности почек.
Острая кишечная инфекция - болезнь грязных рук. Микробы проникают в организм человека при контакте с инфицированными животными или людьми. Заражение возможно при употреблении сырого молока, плохо вымытых фруктов и овощей. Недостаточно эффективная тепловая обработка мяса также становится причиной кишечных расстройств.
Атипичный ГУС
Существует несколько теорий возникновения атипичного ГУС:
- Инфекционная - возбудителями патологии являются микробы: пневмококк, вирус ветряной оспы, ВИЧ, гриппа, Эпштейна-Барра, Коксаки.
- Лекарственная - развитие патологии после приема некоторых медикаментов: антибиотиков, гормональных контрацептивов, цитостатиков.
- Наследственная – выявлены семейные случаи заболевания с аутосомно-доминантным и аутосомно-рецессивным типом наследования.
- Поствакцинальная - развитие ГУС происходит после введения живых вакцин.
аГУС развивается у лиц:
- перенесших обширное хирургическое вмешательство,
- имеющих раковые заболевания,
- страдающих системными заболеваниями – склеродермией, гломерулонефритом,
- имеющих в анамнезе злокачественную гипертонию,
- беременных женщин,
- ВИЧ-инфицированных,
- наркоманов.
Атипичный гемолитико-уремический синдром (АГУС) является осложнением основного заболевания, отличается от типичной формы этиологией, клиникой, гистологической и патоморфологической картиной, неблагоприятным прогнозом и высоким уровнем летальности. Патология развивается в 10% случаев.
Патогенез
Типичный ГУС
Патогенетические звенья типичного гемолитико-уремического синдрома:

Бактериальные токсины поражают клетки кишечника, что приводит к развитию геморрагического колита. В легких возникает дистресс-синдром. Почечная ткань очень чувствительна к действию бактериальных токсинов. Нарушение фильтрационной функции почек приводит к накоплению продуктов обмена, развитию уремии и интоксикации организма. Микроциркуляторные метаморфозы во внутренних органах обуславливают симптоматику ГУС.

Атипичный ГУС
Патогенетические звенья атипичного ГУС:
- гиперактивность и дисфункция системы комплимента,
- образование иммунных комплексов и их отложение на эндотелии сосудов,
- повреждение эндотелиальных клеток и их разрушение,
- ишемия тканей,
- нарушение работы внутренних органов.
У здоровых людей система комплимента разрушает патогенные биологические агенты - бактерии и вирусы. При гемолитико-уремическом синдроме повышенная иммунная активность способствует образованию антител, которые «атакуют» собственные клетки организма - эндотелиоциты и уничтожают здоровые органы. Непрерывная активация тромбоцитов приводит к тромбообразованию и сбою в работе пораженных органов.
Симптоматика
Периоды гемолитико-уремического синдрома - продрома, разгар, восстановление.
Продромальный период длится 2-7 суток и начинается с появления неспецифической симптоматики - недомогания, слабости, катаральных явлений. Кишечные симптомы представлены проявлениями гастроэнтероколита: поносом с кровью, диспепсией, болью в животе. Респираторные клинические признаки - ринит, боль и першение в горле, чихание, кашель.

В разгар болезни общее состояния пациентов ухудшается, повышенная возбудимость, беспокойство и судорожная готовность сменяются вялостью, апатией. Больные дети не кричат и не плачут, они слабо реагируют на внешние раздражители. Возникают симптомы анемии, тромбоцитопении и дисфункции почек. В организме развивается обезвоживание, нарушается периферическое кровообращение. У трети пациентов развивается внепочечный тромбоз.
- Кожа становится бледной, склеры иктеричными, веки пастозными. По мере нарастания гемолитических процессов бледность кожи сменяется желтушностью.
- Геморрагический синдром проявляется носовыми кровотечениями, петехиями или экхимозами на коже и слизистой, кровоизлиянием в стекловидное тело или сетчатку глаза.
- Симптомами почечного синдрома являются олигурия или анурия, гематурия, протеинурия.
- Неврологические нарушения - угнетение сознания, прогрессирующая вялость, нервный тик, нистагм, атаксия, судорожный синдром, ступорозное состояние, кома.
- Поражение сердца и сосудов - учащенное сердцебиение, приглушенность тонов, систолический шум, экстрасистолия, признаки кардиомиопатии, инфаркта миокарда, гипертонии, диффузной васкулопатии, сердечной недостаточности.
- Поражение бронхо-легочной системы - одышка, жесткое дыхание, мелкопузырчатые хрипы.
- Симптомы поражения ЖКТ - отрыжка, изжога, горечь во рту, тошнота, рвота, боль в животе, метеоризм, урчание, неустойчивый стул, патологические примеси в кале.
Если своевременно начать патогенетическую терапию, то олигоанурическая стадия сменится полиурической. Организм больного начнет катастрофически быстро терять воду и электролиты. После разгара заболевания наступает следующая стадия - восстановление. Общее состояние больных стабилизируется, нарушенные функции постепенно нормализуются. У пациентов в крови увеличивается содержание тромбоцитов, улучшается мочеотделение, нормализуется уровень гемоглобина и уменьшается интоксикация.

Среднетяжелое течение гемолитико-уремического синдрома характеризуется быстрым прогрессированием почечной дисфункции: нарастанием интоксикации, анурией, возникновением внепочечных патологий, лихорадкой. В запущенных случаях формируется декомпенсация почечной недостаточности и летальный исход.
Симптомы атипичного ГУС аналогичны клиническим проявлениям его типичной формы. Заболевание отличается бурным течением и развитием серьезных осложнений. Диагноз ставят, если нет ассоциации с диареей и отсутствует лабораторное подтверждение наличия шига-токсина в организме.
Диагностика
Диагностика ГУС основывается на жалобах больных, данных осмотра и результатах лабораторных испытаний.

- В общем анализе крови больных обнаруживают снижение уровня эритроцитов и тромбоцитов. Красные кровяные тельца фрагментируются и приобретают искаженную форму палочек или треугольников.
- В биохимическом исследовании крови определяют повышенное содержание мочевины, креатинина, билирубина, трансаминаз, калия, магния, остаточного азота, снижение белка, хлора и натрия. Такое соотношение микроэлементов указывает на нарушение водно-электролитного баланса и развитие дегидратации в организме.
- В моче определяют много белка и эритроцитов.
- В результате микробиологического исследования кала выявляют энтерогеморрагическую кишечную палочку в значительном количестве, в копрограмме – эритроциты.
Лечение
Лечение гемолитико-уремического синдрома у детей проводится в условиях стационара. Больным показан постельный режим и диетотерапия. Детям назначают диету, разрешающую употреблять только грудное молоко и молочнокислые смеси. Рацион расширяют постепенно. Взрослым рекомендуют соблюдать диету, ограничивающую использование соли.

- Этиотропная терапия - противомикробная. Больным проводят антибиотикотерапию с помощью препаратов широкого спектра действия. В настоящее время широко используют стафилококковый, сальмонеллезный, клебсиеллезный и прочие бактериофаги.
- Патогенетическая терапия заключается в устранении и предупреждении процессов тромбообразования. Больным назначают антикоагулянты и антиагреганты – «Курантил», «Гепарин», а также препараты, улучшающие микроциркуляцию крови – «Трентал», «Кавинтон», «Пирацетам», «Винпоцетин».
- Дезинтоксикационная терапия - введение коллоидных и кристаллоидных растворов.
- Антиоксидантная терапия витамином Е.
- Заместительная терапия – переливание свежезамороженной плазмы, эритроцитарной массы.
- Во время олигоурического периода назначают мочегонные средства – «Фуросемид», «Лазикс»; проводят плазмаферез, перитонеальный диализ или гемодиализ.
- При тахикардии и гипертензии – адреноблокаторы: «Атенолол», «Бетапролол» и ингибиторы АПФ: «Капотен», «Анаприл».
- При отеке легких – «Эуфиллин», искусственная вентиляция легких.
Гемолитико-уремический синдром - тяжелая патология, имеющая серьезный прогноз. У маленьких детей часто наступает смертельный исход, у подростков и взрослых лиц развивается почечная недостаточность и снижается клубочковая фильтрация. Типичный ГУС протекает намного легче недиарейного синдрома, который отличается частым рецидивированием и высоким уровнем смертности.
Профилактика типичной формы ГУС заключается в выполнении некоторых правил. Специалисты рекомендуют:
- соблюдать правила личной гигиены,
- не купаться в сомнительных водоемах,
- пить только кипяченую воду,
- придерживаться технологии обработки и приготовления мясных блюд,
- не употреблять сырое молоко,
- хорошо мыть овощи и фрукты,
- избегать контакта с людьми, страдающими острой кишечной инфекцией.
Видео: презентация по гемолитико-уремическому синдрому
— это чрезвычайно редкое (орфанное) заболевание, связанное с . По системе МКБ-10 кодируется, как D 59.3
Атипичный гемолитико-уремический синдром (аГУС). Прогноз. Причины.
К сожалению, прогноз заболевания, чаще всего, неблагоприятный. Атипичный гемолитико-уремический синдром (аГУС) – это угрожающая жизни хроническая болезнь, характеризующаяся мультисистемным поражением органов и тканей. Заболевание примерно одинаково встречается как у детей, так и у взрослых. В основе заболевания лежит тромботическая микроангиопатия (TMA), при которой в маленьких кровеносных сосудах возникают тромбы. Из-за этого нарушается кровоснабжение, и органы страдают.
 Причина болезни – сбой в системе комплемента, его очень активная деятельность вместо того, чтобы защищать организм, начинает его разрушать. Контролируют уровень активность особые белки, при атипичным гемолитико-уремический синдроме (аГУС) их функция значительно нарушена, и процесс выходит из под контроля.
Причина болезни – сбой в системе комплемента, его очень активная деятельность вместо того, чтобы защищать организм, начинает его разрушать. Контролируют уровень активность особые белки, при атипичным гемолитико-уремический синдроме (аГУС) их функция значительно нарушена, и процесс выходит из под контроля.
Чем дольше длится токсическое агрессивное воздействие на организм, тем более пагубными становятся последствия: отказывают почки, развивается инсульт или инфаркт.
Соответственно, при ранней диагностике, при выявлении заболевания на начальных стадиях, можно снизить пагубное воздействие атипичного гемолитико-уремического синдрома (аГУС), и тем самым дать шанс человеку на нормальную полноценную жизнь.
Атипичный гемолитико-уремический синдром (аГУС). Наследование.
Атипичный гемолитико-уремический синдром (аГУС) считается наследственным заболеванием только в 20% случаев, с аутосомно-рецессивным или доминантным типом передачи. Примерно в половине случаев выявить генетическую мутацию не получается. Поэтому ДНК – анализ при атипичном гемолитико-уремическом синдроме (аГУС) не является важнейшим диагностическим методом, на его основании не может даваться заключение о начале или отмене терапии.
Атипичный гемолитико-уремический синдром (аГУС) одинаково распространен по всей планете, не выявлено зависимости от пола или расы пациентов. Из-за редкости заболевания сложно говорить о точном количестве пациентов, есть предположения о том, что болезнь встречается примерно от 1 до 9 случаев на миллион человек.
Атипичный гемолитико-уремический синдром (аГУС). Клинические проявления.
Симптомы атипичного гемолитико-уремического синдрома (аГУС) встречаются как все вместе, так и по отдельности. Насторожить должно любое из клинических проявлений болезни.
Итак, симптомами атипичного гемолитико-уремического синдрома (аГУС) являются:
- Постоянная усталость и недомогание;
- Поражение почек вплоть до необходимости гемодиализа из-за терминальной почечной недостаточности;
- Отеки, тяжесть в ногах;
- Снижение диуреза;
- Повышение креатинина крови;
- Снижение скорости клубочковой фильтрации;
- Артериальная гипертензия;
- Внепочечный тромбоз;
- Протеинурия;
- Энцефалопатия;
- Спутанность сознания;
- Судороги;
- Инсульт;
- Инфаркт;
- Кардиомиопатия с развитием сердечной недостаточности;
- Высокое артериальное давление;
- Поражение глазных сосудов;
- Поражение легких;
- Поражение кожи. Сыпь;
- Боли в животе;
- Диарея;
- Рвота;
- Колит;
- Панкреатит.
Вызывать проявления атипичного гемолитико-уремического синдрома (аГУС) могут несколько причин, зачастую они становятся спусковым крючком для начала заболевания. Вот они:
- Диарея;
- Гастроэнтерит;
- Инфекции верхних дыхательных путей;
- Беременность и роды;
- Гломерулопатия;
- Склеродермия;
- Системная красная волчанка;
- Злокачественная артериальная гипертензия;
- Злокачественные новообразования;
- Трансплантация почки и костного мозга.
Все они усиливают нарушение в системе комплемента. Иногда для манифестации заболевания бывает достаточно одного фактора, но они могут действовать и в совокупности.
Атипичный гемолитико-уремический синдром (аГУС). Диагностика.
 Тромботическая микроангиопатия (TMA) – результат атипичного гемолитико-уремического синдрома (аГУС), но вызвать такое тяжелое последствие может не только аГУС, но и другие заболевания. Поэтому так важно проводить дифференциальную диагностику, чтобы квалифицировать именно манифестицию атипичного гемолитико-уремического синдрома (аГУС).
Тромботическая микроангиопатия (TMA) – результат атипичного гемолитико-уремического синдрома (аГУС), но вызвать такое тяжелое последствие может не только аГУС, но и другие заболевания. Поэтому так важно проводить дифференциальную диагностику, чтобы квалифицировать именно манифестицию атипичного гемолитико-уремического синдрома (аГУС).
Во-первых, атипичный гемолитико-уремический синдром (аГУС) не имеет в самом начале возникновения заболевания проявлений гемоколита, во-вторых диарея может сама по себе вызвать заболевание, а не быть его симптомом. На этом этапе нужно исключить STEC и Streptococcus pneumoniae – инфекции.
Дальше методом исключения проверяем пациента на системную красную волчанку, СПИД, злокачественную гипертонию, тромботическую тромбоцитопеническую пурпуру, HELLP — синдром у рожающих женщин и ряд других заболеваний.
Напоминаем, что генетический анализ не имеет 100% достоверности, у части пациентов с подтвержденным атипичным гемолитико-уремическим синдромом (аГУС), нарушения в соответствующем гене не выявлены.
Атипичный гемолитико-уремический синдром (аГУС). Лечение.
Лечение инфузиями плазмы, пламообменом. В настоящий момент метод считается недостаточно эффективным, у одних пациентов он вызывает незначительное улучшение показателей, а у других оказывается практически бесполезным.
Гемодиализ. Процедура искусственной очистки организма лишь устраняет последствия сбоя в системе комплемента, но при этом никак не влияет на процесс избыточного образования белка. Она способна на несколько лет увеличить продолжительность жизни пациента. Сохраняется возможность развития внепочечной тромботической микроангиопатии.
Трансплантация. Почка, потерявшая свою функцию, может быть заменена на новую, но прогрессирующее заболевание может вновь начать разрушать органы и ткани. 90% пациентов опять чувствуют симптомы атипичного гемолитико-уремического синдрома (аГУС). Есть большой риск рецидива после трансплантации. Иногда можно осуществить двойную трансплантацию, и печени, и почки, но при этом возникает огромная сложность в поиске идеально подходящих двух донорских органов. Кроме того, и такая сложнейшая манипуляция может не дать гарантированного положительного результата. Тромботическая микроангиопатия развивается и в других органах.
 Экулизумаб.
Единственный препарат, который воздействует на механизм развития заболевания, а не на последствия болезни. Экулизумаб связывает компонент комплемента и тем самым предотвращает накопление негативных факторов. Препарат зарегистрирован в России и доказал свою эффективность. Люди с атипичным гемолитико-уремическим синдромом (аГУС) не чувствуют постоянную усталость, у них не развивается поражение органов и тканей. Внедрение в лечебную практику экулизумаба, гуманизированного моноклонального антитела к С5 — фракции терминальной стадии каскада комплемента, значительно увеличивает продолжительность жизни пациентов, а сама жизнь становится полноценной.
Экулизумаб.
Единственный препарат, который воздействует на механизм развития заболевания, а не на последствия болезни. Экулизумаб связывает компонент комплемента и тем самым предотвращает накопление негативных факторов. Препарат зарегистрирован в России и доказал свою эффективность. Люди с атипичным гемолитико-уремическим синдромом (аГУС) не чувствуют постоянную усталость, у них не развивается поражение органов и тканей. Внедрение в лечебную практику экулизумаба, гуманизированного моноклонального антитела к С5 — фракции терминальной стадии каскада комплемента, значительно увеличивает продолжительность жизни пациентов, а сама жизнь становится полноценной.
Гемолитико-уремический синдром (ГУС) представляет собой серьезную терапевтическую проблему в педиатрии и детской нефрологии, являясь одной из ведущих причин острой почечной недостаточности с потенциальной трансформацией в терминальную хроническую почечную недостаточность в различные сроки от начала заболевания.
Несмотря на то, что наиболее распространена типичная форма ГУС с диарейной продромой, ассоциированная с токсином Шига (STEC), требуется тщательное подтверждение инфекционной этиологии, чтобы вовремя исключить атипичный ГУС и ГУС, связанный с пневмококковой инфекцией. В отношении STEC-ГУС рекомендуется адекватная симптоматическая терапия со своевременным подключением диализа при необходимости. Прогноз при этом будет зависеть от продолжительности анурического периода и сопутствующих повреждений центральной нервной системы.
В соответствии с рекомендациями Инициативы по улучшению глобальных исходов заболеваний почек (Kidney Disease: Improving Global Outcomes, KDIGO) их сила указана как уровни «1», «2» или «нет градации», качество доказательной базы - как А, В, С, D.
Типичный гемолитико-уремический синдром (ГУС)
- При развитии острой почечной недостаточности в возрасте от 6 мес до 3 лет велика вероятность ГУС в качестве ее причины.
- Аргументом в пользу ГУС может служить анамнестическое указание на предшествующий эпизод диареи с примесью крови в стуле.
- Клиническими признаками ГУС, помимо симптомов острой почечной недостаточности (олигурия, азотемия, гипергидратация и др.), являются Кумбс-негативная гемолитическая анемия с присутствием в мазке шизоцитов и тромбоцитопения, отражающие активный процесс тромботической микроангиопатии.
- В качестве необходимого диагностического теста рекомендуется исследование Шига-токсина в стуле методом полимеразной цепной реакции или определение IgM-антител к липополисахариду Escherichia coli, продуцирующей Шига- токсин, являющийся основным этиологическим фактором ГУС (1A).
- При развитии острой почечной недостаточности с анурией, а также некорригируемыми гипергидратацией, электролитными расстройствами и артериальной гипертензией рекомендуется незамедлительная инициация заместительной почечной терапии (перитонеальный диализ, гемодиализ, продленная вено-венозная гемодиафильтрация) с учетом возраста и состояния гемодинамики больного (1B).
- Не рекомендуется назначение антибиотиков для лечения диареи при ГУС с учетом возможного усиления циркуляции Шига-токсина из разрушенных микробных клеток (2B).
- Для коррекции выраженной анемии с симптомами гипоксемии рекомендуются трансфузии эритроцитарной массы (1A).
- За исключением выраженного кровотечения, не рекомендуется введение тромбоцитарной массы, так как это может привести к усилению образования микротромбов (2B).
- Дети, перенесшие ГУС, нуждаются в длительном наблюдении с учетом вероятности отдаленных последствий в виде формирования хронической болезни почек.
Атипичный гемолитико-уремический синдром (аГУС).
- Отсутствие предшествующей диареи, негативный результат исследования на Шига-токсин, семейный и рецидивирующий характер заболевания, признаки активации альтернативного пути комплемента, мультиорганность поражения требуют исключения аГУС - комплементопос- редованной тромботической микроангиопатии (ТМА), чаще всего вызванной мутациями генов белков системы комплемента.
- При подозрении на аГУС рекомендуется исследование активности фактора ADAMTS13 для исключения тромботической тромбоцитопенической пурпуры (1A).
- Дифференциальную диагностику аГУС следует проводить с ТМА при системной красной волчанке, применении ряда лекарственных препаратов и метилмалоновой ацидурии (1B).
- Рекомендуется исследование уровня антител к фактору Н комплемента (CFH) для исключения антителоопосредованной формы аГУС (1B).
- В качестве патогенетической терапии аГУС рекомендуется применение экулизумаба - моноклонального антитела к С5 компоненту комплемента, блокирующего дистальную часть альтернативного пути его активации (1B).
- При выявлении высокого уровня антител к фактору Н возможно использование иммуносупрессивной терапии ритуксимабом (2B).
- При невозможности незамедлительного начала лечения экулизумабом рекомендуется плазмотерапия в виде плазмообменов или трансфузий свежезамороженной плазмы (2B).
- Для определения продолжительности терапии экулизумабом рекомендуются оценка его эффекта на протяжении нескольких месяцев и проведение молекулярно-генетического исследования для выявления мутаций генов системы комплемента: CFH, CFI, CFB, C3, THBD, MCP (2B).
Введение
Гемолитико-уремический синдром является одной из наиболее частых причин развития острой почечной недостаточности у детей; характеризуется триадой симптомов: Кумбс-негативной гемолитической анемией с наличием фрагментированных эритроцитов (шизоцитов), тромбоцитопенией и острой почечной недостаточностью.
Указанные признаки являются составляющими тромботической микроангиопатии - генерализованной окклюзии сосудов мелкого калибра тромбами, возникшими вследствие повреждения эндотелия. В результате поражения эндотелиальных клеток происходит механическое повреждение эритроцитов, активация агрегации тромбоцитов с образованием тромбов в микроциркуляторном русле, особенно в почках.
У детей раннего возраста в большинстве случаев (90-95%) развивается так называемый типичный или постдиарейный ГУС (Д + ГУС), который вторичен по отношению к инфекции Escherichia coli, продуцирующей так называемый Шига-токсин (Shigatoxine - Stx, продуцирующий E. coli; STEC). Реже инфекционными стимулами служат шигеллы и пневмококки. Другая форма ГУС, называемая атипичной, встречается гораздо реже (5-10% всех случаев) и является результатом аномалии (чаще генетической) белков, регулирующих процесс активации комплемента.
Типичный постдиарейный гус
Д+ ГУС как следствие STEC-инфекции является наиболее частой формой ГУС у детей. Отмечается в основном в возрасте до 3 лет и редко - до 6 мес. Частота составляет ~2-3 случая на 10 000 детей в возрасте до 3 лет.
Патогенез
STEC-инфекция обнаруживается приблизительно в 85% случаев Д+ ГУС с помощью посева стула или ректального мазка в питательную среду Мак-Конки (Mac Conkey) с сорбитолом. Наиболее часто встречающимся серотипом является 0157:H7 (реже O111, O103, 0121 и др.). Другими вариантами диагностики STEC-инфекции являются обнаружение гена Шига-токсина в стуле методом полимеразной цепной реакции, или, реже, определение IgM-антител к липополисахариду наиболее часто встречающихся серогрупп микроорганизма в сыворотке крови.
Резервуаром инфекции являются кишечник и фекалии крупного рогатого скота. Возможными переносчиками могут быть также овцы, козы, лошади, олени, кошки, собаки, птицы и мухи. Человек заражается при употреблении полусырой рубленой говядины, непастеризованного некипяченого молока, сыра, фруктов, соков, овощей, зараженной воды из колодца и водоемов, а также при неисправности водопровода. Прямой контакт детей с животными или их испражнениями и передача от человека к человеку являются другими серьезными источниками заражения.
Д+ ГУС может быть спорадическим либо, в случае заражения из одного и того же источника, манифестировать с промежутком в несколько дней или недель у сибсов. Часто члены семьи имеют STEC-диарею без развития ГУС.
Эпидемии диареи или геморрагического колита в результате инфицирования STEC из единого источника, охватывавшие сотни людей, отмечены в различных странах, частота ГУС среди них составила 10-20%.
Патогенетическая связь между кишечной контаминацией STEC и ГУС не полностью ясна. Микроорганизм прикрепляется к ворсинкам слизистой оболочки толстой кишки и выделяет Шига-токсин. Остается неясным, каким образом Шига-токсин перемещается из кишечника к своей цели - эндотелиальным клеткам сосудов. Транспортерами Шига-токсина могут быть полинуклеарные нейтрофилы, моноциты или тромбоциты. Токсин прикрепляется к своему рецептору (глоботриаосилцерамиду, Gb3) на сосудистых эндотелиальных клетках почек, центральной нервной системы (ЦНС) и других органов.
После связывания с Gb3 активная часть Шига-токсина проникает в клетку, подавляя синтез белков, что, в свою очередь, приводит к смерти клеток эндотелия. Шига- токсин индуцирует также местную продукцию цитокинов, которые запускают каскад воспалительных и прокоагуляционных событий.
Клиническая картина
В продромальной фазе Д+ ГУС отмечаются диарея (у 90-95%), рвота (у 30-60%) и боли в животе. В 70% случаев через 1-2 дня от начала заболевания в стуле появляется кровь. Манифестация ГУС начинается в среднем через 6 (2-14) дней. Бледность кожных покровов, общее недомогание, слабость, летаргия, изменение поведения, небольшая желтушность, уменьшение количества мочи после «кровавой» диареи должны насторожить врача в отношении ГУС.
ГУС начинается внезапно и характеризуется типичной комбинацией признаков.
- Гемолитическая анемия: уровень гемоглобина, как правило, < 80 г/л, имеются шизоциты (2-10%); 70% пациентов нуждаются в трансфузии крови.
- Тромбоцитопения (~ 50 000-70 000х10*9/л) не является достаточно выраженной, чтобы вызвать кровотечения в отсутствии хирургических вмешательств, хотя у некоторых детей появляется кожный геморрагический синдром.
- Лейкоцитоз более 20,0х10*9/л в тяжелых случаях ГУС является частой находкой.
- Острая почечная недостаточность с повышением уровня сывороточного креатинина и мочевины. Приблизительно половина пациентов имеют тяжелую олигурию или анурию, 50-60% нуждаются в остром диализе.
При наличии мочи постоянно определяются микро- или макрогематурия и протеинурия. Поскольку анурия диагностируется с опозданием, у пациентов прогрессирует гипергидратация, поэтому первыми проявлениями ГУС могут быть гипонатриемия и гиперволемия с артериальной гипертензией. В других случаях отмечается обезвоженность из-за диареи и рвоты. Уровень сывороточного калия, который сначала может быть низким из-за кишечных потерь, быстро повышается. Часто отмечаются ацидоз, гипокальциемия, гиперфосфатемия и гиперурикемия.
Экстраренальные проявления
- Поражение центральной нервной системы, которое является основной причиной смерти, отмечается ~ у 20% детей: частыми симптомами являются фокальные или генерализованные судороги, стридор, нарушение сознания; возможны гемипарестезия или гемиплегия, корковая слепота, кома, иногда децеребрация с вовлечением ствола головного мозга.
Сначала результаты компьютерного или магнитно-резонансного сканирования могут быть нормальными или выявлять участки пониженной плотности. В случае ограниченного и обратимого ишемического поражения возможно полное восстановление нервной системы. Диффузные либо локализованные в стволе мозга некротические изменения могут привести к смерти или тяжелым неврологическим последствиям.
- Серьезное поражение желудочно-кишечного тракта (~ у 10%): тяжелый геморрагический колит с постоянной меленой, боль, рвота, состояние частичной непроходимости; реже токсический мегаколон, инвагинация, перфорация толстой кишки или выраженный некроз, вторичный стеноз толстой кишки.
- Отек поджелудочной железы при ультразвуковом исследовании в сочетании с повышением уровня амилазы и липазы (~10% пациентов). Редко развивается некротизирующий панкреатит. В результате некроза островковых клеток возможно развитие транзиторного или перманентного инсулинзависимого сахарного диабета.
- Поражение печени (у 40%): проявляется гепатомегалией, повышением уровня трансаминаз и имеет относительно доброкачественное течение.
- Сердечно-сосудистые осложнения (за исключением сердечной недостаточности в результате гиперволемии и гипертензии) встречаются редко (2%) и включают ишемию миокарда с сердечной недостаточностью, аритмии, миокардит или тампонаду сердца.
Прогноз
Смертность в 2000-е гг. (в основном в результате поражения ЦНС) составила 1-5%.
В большинстве случаев в течение < 1-2 нед исчезают гемолитическая анемия и тромбоцитопения, нормализуется диурез. Несмотря на это, в среднем в течение 4 мес катамнестического наблюдения ~10% детей достигают терминальной хронической почечной недостаточности (ХПН). ХПН иногда развивается уже в острой стадии после транзиторного частичного улучшения функции почек. В дополнение у 25% детей отмечаются остаточные изменения почек: снижение клубочковой фильтрации (70-80 мл/1,73 м2 в мин), гипертензия или протеинурия.
К факторам риска перманентного поражения почек в острой стадии относятся необходимость в гемодиализе более 5 дней, длительность олигоанурии более 10 дней, количество полинуклеаров > 20,0х10 э/л, поражение ЦНС, тяжелые кишечные осложнения. У большинства пациентов этой группы через 20-30 лет развивается терминальная ХПН.
Ведение ребенка с Д + ГУС
Крайне необходимо своевременное подтверждение факта STEC-инфекции и определение основных признаков тромботической микроангиопатии, показателей азотемии, электролитов и основных витальных параметров. При развитии олигурии следует предусмотреть возможность начала диализа.
Коррекция водно-электролитного обмена
Необходим расчет жидкости с ее ограничением при гипергидратации и, наоборот, компенсацией потерь со стулом, рвотой и сохраненном диурезе, так как дегидратация может усугубить ишемическое повреждение почек и других органов. Признаками гипергидратации могут быть увеличение массы тела, артериальная гипертензия, отеки, гипонатриемия.
Попытки применения высоких доз фуросемида (2-5 мг/кг) редко позволяют достичь эффекта, равно как и гипотензивная терапия периферическими вазодилататорами, поэтому предпочтение отдается диализу, особенно при наличии выраженной гиперкалиемии и метаболического ацидоза, коррекция которых введением растворов бикарбоната и глюкозы может усугубить гипергидратацию.
Питание
Питание так же, как воду и электролиты, лучше обеспечивать перорально, при необходимости - через желудочный зонд. Количество калорий и белка должно составить 100% рекомендованной суточной потребности. Необходимость в парентеральном питании возникает в случае продолжающихся рвоты диареи и симптомов колита.
Переливание крови
Эритроцитарную массу вводят при уровне гемоглобина ниже 70 г/л. С целью предотвращения анти-HLA- иммунизации трансфузию рекомендуется проводить через специальные фильтры (задерживающие лейкоциты и тромбоциты). При отсутствии серьезных кровотечений и показаний к инвазивным мероприятиям (установление центрального или перитонеального катетера, абдоминальные хирургические вмешательства) нет необходимости в введении тромбоцитарной массы. Более того, введение тромбоцитов может усугубить процесс тромбообразования.
Диализ
Необходимость диализа определяется в первую очередь наличием или отсутствием олигурии. Диализ (обычно перитонеальный с помощью катетера Tenckhoff) желательно начать до развития осложнений острой почечной недостаточности.
Терапия осложнений
Дети даже с умеренными неврологическими симптомами нуждаются в пристальном наблюдении и частых исследованиях, нередко в отделении интенсивной терапии: ухудшение может развиться стремительно.
Для обеспечения своевременного оперативного вмешательства при перфорации/некрозе кишечника или вторичном стенозе в ведении пациента должен участвовать хирург. При наличии сахарного диабета необходима инсулинотерапия. У детей с кардиомегалией и сердечной недостаточностью рекомендуется тщательный мониторинг сердечной деятельности.
Специфическая терапия
Нет доказанного варианта терапии, способного повлиять на течение Д+ ГУС. Гепарин, тромболитики и антиагреганты, стероиды и свежезамороженная плазма (СЗП) не имеют существенного эффекта. В тяжелых случаях, особенно при поражении ЦНС, проводят заменное переливание плазмы (ЗПП). Целью является удаление факторов свертывания, тромбообразования и замещение с помощью СЗП потенциально полезных веществ, главным образом антитромбинов.
Трансплантация почки
Риск развития возвратного Д+ ГУС после трансплантации почки отсутствует. Необходимо обсудить возможность трансплантации от живого родственного донора. Циклоспорин не противопоказан. На основании анализа течения заболевания должен быть исключен атипичный ГУС, при необходимости - путем молекулярно-генетического исследования.
Предотвращение инфицирования STEC и профилактика развития ГУС
Следует ознакомить родителей маленьких детей с правилами предотвращения контаминации STEC:
- рубленая говядина должна быть хорошо прожарена до появления серого цвета на разрезе;
- дети до 3 лет не должны употреблять непастеризованные продукты (молоко, сыр, фруктовые соки);
- до приготовления пищи, особенно после манипуляций с рубленой говядиной, необходимо мыть руки;
- дети, которые прикасались к крупному рогатому скоту и другим животным, должны вымыть руки и умыться после этого, а также перед едой;
- для предотвращения контаминации мяса кишечным содержимым необходим контроль убоя скота. Важен надлежащий надзор и уход за системой водоснабжения;
- антибиотики: многочисленными исследованиями показано, что антибиотикотерапия в период диареи увеличивает риск развития ГУС, возможно, в связи с освобождением Шига-токсина в результате лизиса бактерий. Тем не менее этот риск пока не доказан. Необходимо также уточнить, стоит ли назначать антибиотики, не вызывающие бактериальный лизис, такие как макролиды (азитромицин), сибсам пациентов с STEC-позитивным ГУС.
ГУС в результате инфекции Shigella dysenteriae тип 1
S.dysenteriae тип 1, продуцирующий Шига-токсин, является основной причиной ГУС в эндемичных регионах, таких как Бангладеш или Африка. Этот тип ГУС протекает тяжелее, чем STEC-ГУС. В 20% случаев отмечается бактериемия, часто с развитием септического шока и внутрисосудистой коагуляции. Смертность колеблется в пределах 20-40%. У 40% развивается ХПН, которая в течение нескольких лет достигает терминальной стадии. Раннее назначение антибиотиков (цефалоспорины 3-го поколения или хинолоны) снижает риск развития ГУС у детей, инфицированных S. dysenteriae тип 1.
ГУС, вторичный по отношению к Streptococcus pneumoniae
Выделяют особую форму ГУС, которая развивается непосредственно после инфекции S. pneumoniae (пневмония и/или эмпиема и менингит), в основном у детей в возрасте младше 2 лет.
Механизм развития этой формы ГУС особенный. Нейраминидаза S. pneumoniae атакует N-ацетил-нейраминовую кислоту поверхности клеток, обнажая при этом холодовой T-антиген (криптантиген; Thomsen- Friedenreich antigen) - компонент клеточных мембран эритроцитов, тромбоцитов, эндотелиальных клеток клубочков. У человека имеются естественные антитела к Т-антигену, которые приводят к агглютинации эритроцитов и запуску процессов, приводящих в итоге к развитию ГУС. При пневмококковой инфекции положительный тест на Т-активацию свидетельствует о повышенном риске развития ГУС. Прямой тест Кумбса обычно также позитивен. Смертность, в основном обусловленная менингитом, составляет ~10%. Другие 10% пациентов быстро развивают терминальную почечную недостаточность; 20% имеют остаточные явления - нарушение почечной функции, гипертензию.
Введение плазмы и неотмытых эритроцитов противопоказано, поскольку они содержат анти-Т IgM-антитела, которые могут спровоцировать рецидив ГУС. Отдельные работы свидетельствуют об эффективности ЗПП с последующим замещением альбумином.
Атипичный гус
Общепризнанного определения аГУС не существует. Одно из имеющихся гласит, что аГУС - это ГУС без сопутствующей болезни. Под сопутствующей болезнью понимают гемоколит, вызванный STEC-инфекцией, пневмококковую пневмонию, системную красную волчанку, тромботическую тромбоцитопеническую пурпуру, наследственные нарушения обмена кобаламина, патогенное воздействие лекарств и другие патологические состояния, способные вызвать ТМА. В более узком и традиционном понимании аГУС - это ГУС, опосредованный дисфункцией системы регуляции комплемента с неконтролируемой активацией его альтернативного пути.
Атипичный вариант составляет 5-10% всех случаев ГУС у детей и в основном является следствием нарушения регуляции системы комплемента. Отдельные редкие случаи у младенцев (метилмалоновая ацидемия) - это результат наследственной аномалии внутриклеточного метаболизма кобаламина (витамин B12). По данным различных исследований, распространенность аГУС колеблется от 1 до 7 случаев на 1 000 000 населения.
Патогенез
Система комплемента является основным фактором защиты от микроорганизмов. При нормальной регуляции активация комплемента специфически направлена на поверхность микроба, однако подавляется на поверхности клеток хозяина. При активации комплемента образуется конвертаза C3bBb, которая приводит к превращению C3 в C3b.
В результате происходит отложение C3b на поверхности микробов (опсонизация) и формирование мембраноатакующего комплекса (МАК, или C5b9), который приводит к лизису микробной клетки. На поверхности клеток хозяина этот процесс строго контролируется белками-регуляторами, к которым относятся комплементарный фактор H (CFH), фактор I (CFI) и CD46, или мембранный кофакторный протеин, нециркулирующий протеин, закрепленный на поверхности клеток (MCP). Эти три фактора, действуя сообща, предотвращают активацию и депозицию C3b на клетках.
Мутации этих белков приводят к утрате защиты эндотелиальных клеток от повреждения конечными продуктами активации альтернативного пути комплемента. Иными словами, нарушается процесс подавления избыточной активности системы комплемента, что приводит к реализации повреждающего действия конечных продуктов его альтернативного пути на клетки эндотелия с развитием ТМА.
аГУС ассоциируется с мутациями CFH у 20-25% пациентов, MCP — 15% и CFI — 10%. Мутации фактора В (CFB) встречаются крайне редко (1%), в то время как мутации C3 фракции комплемента - у 10% пациентов. Редкими являются мутации гена тромбомодулина (THBD). Приблизительно 10% детей имеют сочетанные мутации. В дополнение 10% детей имеют приобретенный функциональный дефицит CFH в связи с наличием анти- CFH антител. Только 30% заболеваний аГУС не находит сегодня должного объяснения с позиций молекулярной генетики.
Диагностика
Исходя из определения аГУС, для постановки его диагноза у ребенка должны быть исключены прочие причины развития ТМА, в первую очередь Д+ ГУС (постдиарейный). В ряде случаев в дебюте аГУС также отмечается диарея, поэтому необходимо своевременное исключение STEC- инфекции, равно как и инфекции S. pneumoniae.
Помимо этого, следует исключить системную красную волчанку, ВИЧ-инфекцию, инфекцию вирусом H1N1, предшествующую злокачественную гипертонию, HELLP- синдром у рожениц, прием лекарств (циклоспорин А), метилмалоновую ацидурию как возможные причины ТМА.
Особое внимание следует уделить исключению тромботической тромбоцитопенической пурпуры (ТТП). Необходимо исследование уровня фактора ADAMTS13, ответственного за инактивацию фактора Виллебранда у всех больных с картиной ТМА, выраженный дефицит которого (ADAMTS13 < 5% нормы) приводит к определенной форме ТТП. Клинически ТТП и ГУС имеют много сходного. У детей ТТП чаще носит врожденный характер и ассоциируется с наследственным полным дефицитом ADAMTS13. Приобретенные формы в результате наличия анти-ADAMTS антител у детей встречаются исключительно редко. Для ТТП характерно превалирование неврологической симптоматики при умеренном нарушении функции почек.
Поскольку многие мутации скорее ведут к нарушению функции белков системы комплемента, нежели к изменению их плазматической концентрации, уровень CFH, CFI, C3 и CFB может оставаться нормальным даже при наличии мутаций. Сам же молекулярно-генетический анализ требует значительного времени, и получение его результатов в острой фазе заболевания практически нереально. Вместе с тем он крайне желателен в процессе наблюдения за больными для определения стратегии их долгосрочной терапии.
Клинические проявления
Очень ранее начало (даже в период новорожденности) характерно для аГУС, связанного с мутациями CFH и CFI (средний возраст 6 и 2 мес, соответственно).
Наоборот, при мутации MCP заболевание всегда начинается после 1 года жизни. Варианты аГУС с неидентифицированными мутациями могут начаться в любом возрасте. Анти-CFH антитела чаще отмечаются ближе к подростковому периоду.
Некоторые инфекции (верхних дыхательных путей, лихорадка, гастроэнтериты) запускают первый эпизод ГУС и рецидивы, независимо от генетического варианта, у 2/3 пациентов. Диарея провоцирует аГУС у 1/3 пациентов, что иногда затрудняет дифференциацию с Д+ ГУС (типичным).
У 1/4 пациентов аГУС носит семейный характер (сибсы, родители, бабушки и дедушки имеют заболевание). Неотягощенный семейный анамнез не исключает возможности генетической передачи заболевания. Лишь половина носителей мутации в семье в течение жизни имеют манифестацию заболевания.
Прогноз
В целом прогноз аГУС неблагоприятный. Смертность в острой стадии составляет 5-10%. Приблизительно у 50% пациентов развивается терминальная ХПН, чаще в течение 1 года от начала манифестации. Экстраренальные проявления, чаще поражение ЦНС (судороги, кома, ишемические очаги при магнитно-резонансной томографии), отмечаются более чем у 20% пациентов.
Рецидивы аГУС отмечаются при всех вариантах, чаще у пациентов с мутацией MCP. Провоцирующие инфекции при этой мутации сопровождаются острым гемолизом, тромбоцитопенией и острой почечной недостаточностью в результате гемоглобинурии. В большинстве этих случаев функция почек полностью восстанавливается. Промежуток времени между рецидивами колеблется иногда от нескольких недель до многих лет.
Наиболее благоприятный прогноз отмечается при MCP, наиболее неблагоприятный - при CFH и сочетанных мутациях. Во французском исследовании смерть или терминальная ХПН в сроки менее 1 года от начала заболевания отмечены у 60% с мутацией CFH, у 37% с мутацией CFI, у 33% с мутацией С3, у 60% с комбинированными мутациями, у 32% в группе с неизвестной этиологией и 0% с мутацией MCP. У больных с анти-CFH антителами в случае раннего лечения плазмообменом заболевание имеет благоприятное течение.
Лечение
Плазма
Введение СЗП долгое время оставалось первой линией терапии. Тем не менее ее эффективность, по данным ретроспективных исследований, не предотвращает развития терминальной ХПН. СЗП является источником нормальных CFH, CFI, C3 и CFB, а также большого количества других функциональных белков. С помощью плазмообмена удаляются мутантные CFH, CFI, C3, CFB и анти-CFH антитела. Предварительное удаление плазмы при ЗПП предотвращает гиперволемию и риск развития сердечной недостаточности в результате введения большого количества СЗП.
Плазмотерапия эффективна в наибольшей степени при мутациях CFH. При мутациях MCP ее эффективность практически отсутствует, так как кодируемый белок экспрессируется на клеточных мембранах, то есть в твердой фазе, а не в циркуляции.
Предпочтительным является мембранный плазмаферез с объемом замещения 50-60 мл на кг массы тела. Альтернативный вариант - инфузии СЗП в объеме 10-20 мл/кг.
Экулизумаб
Терапевтический подход к аГУС был радикально усовершенствован с открытием и внедрением в клиническую практику экулизумаба - гуманизированного моноклонального антитела к С5-фракции терминальной стадии каскада комплемента. Препарат предотвращает расщепление С5, ведущее к образованию провоспалительного С5а и протромботического С5Ь-9 компонентов, тем самым устраняя их патогенное действие. К настоящему моменту опубликованы результаты лечения экулизумабом более 189 пациентов с аГУС; препарат одобрен во многих странах, в том числе в России.
Дополнительное введение препарата рекомендуется при сочетанном применении плазмафереза, так как последний удаляет часть препарата из циркуляции. С учетом единичных сообщений о риске развития менингококкового менингита на фоне применения экулизумаба рекомендуется предварительная вакцинация и/или профилактическое применение антибиотиков.
В опубликованных результатах контролируемых исследований продемонстрирована быстрая ликвидация активности гемолиза (в среднем за 7-14 дней) и признаков активной ТМА у 88% больных с гематологической ремиссией на протяжении всего курса лечения у большинства пациентов. Отмечено повышение скорости клубочковой фильтрации в среднем на 32 мл/1,73 м2 в мин от исходного с наибольшей выраженностью при раннем начале лечения. У детей повышение скорости клубочковой фильтрации было более выраженным (64 мл/1,73 м2 в мин). В ряде случаев наблюдали медленное улучшение почечной функции (на протяжении 2 лет). Большинство пациентов избавились от потребности в диализе.
При сравнении групп больных, получавших и не получавших экулизумаб, отмечена достоверная разница в количественном отношении достигших терминальной стадии ХПН к 1 году наблюдения (25 и 63%, соответственно).
При патогенетической обоснованности пожизненной терапии у носителей мутаций генов системы комплемента длительность терапии экулизумабом остается дискутабельной. В единичном сообщении об отмене экулизумаба у 10 взрослых больных у троих отмечены рецидивы ТМА с прогрессирующим снижением почечной функции.
Поддерживающая терапия
При вариантах аГУС, опосредованных антителами к CFH, помимо терапии экулизумабом, эффективность которой подтверждена, возможно использование иммуносупрессивной терапии. Описаны положительные результаты в отношении гематологических изменений и показателей функции почек на фоне применения пульсовых введений циклофосфамида в дозе 0,5 г/1,73 м2, кортикостероидов и ритуксимаба. Положительная клиническая динамика сопровождалась снижением титра антител к CFH. Для поддерживающей терапии наряду с кортикостероидами применялся микофенолата мофетил.
Профилактика инфекций
Большинство эпизодов аГУС запускаются с помощью инфекций, что обусловливает необходимость эрадикации хронических очагов аденоидной, тонзиллярной и зубной инфекции. У отдельных больных рецидивы отмечены после вакцинации. Тем не менее польза иммунизации существенно превалирует над риском. Мы рекомендуем проведение всех вакцинаций, включая противогриппозную.
Трансплантация почки при aГУС
Среди детей с потребностью в трансплантации почки, возникшей в исходе перенесенного ГУС, доля больных с атипичным вариантом может достигать половины. Риск возврата аГУС сразу после трансплантации чрезвычайно высок у пациентов с мутациями CFH (~80%), CFI и С3 (> 50%). Трансплантация проведена всего 3 пациентам с мутацией CFB: все потеряли почки в результате возвратного аГУС.
Поскольку трансплантированная почка не содержит мутированного MCP-белка, вероятность возврата аГУС при этих мутациях очень мала. Большинство больных при возврате аГУС теряют почки менее чем через 1 год. Другой причиной потери почки у детей является тромбоз.
Лишь у единичных пациентов удалось предотвратить возвратный аГУС путем проведения сеансов ЗПП до операции и в посттрансплантационном периоде. Более оптимистичные перспективы связаны с использованием экулизумаба в до- и послеоперационном периоде трансплантации. Имеются сообщения о 13 пациентах с потерей предыдущего трансплантата, у которых введение экулизумаба за несколько часов до пересадки и в течение 24 ч после нее с постепенным переходом на стандартный режим лечения способствовало предотвращению отторжения и возврата ТМА.
Поскольку CFH, так же, как и CFI, CFB и C3, синтезируются в печени, комбинированную трансплантацию печени и почки либо изолированную трансплантацию печени в случае сохранной функции почек можно рассматривать как вариант терапии. Однако, предварительные результаты показали, что данный метод уступает по эффективности применению экулизумаба.
Заключение
Гемолитико-уремический синдром представляет собой серьезную терапевтическую проблему в педиатрии и детской нефрологии, являясь одной из ведущих причин острой почечной недостаточности с потенциальной трансформацией в терминальную хроническую почечную недостаточность в различные сроки от начала заболевания.
Несмотря на то, что наиболее распространена STEC-ассоциированная форма ГУС с типичной диарейной продромой, требуется тщательное подтверждение инфекционной этиологии для того, чтобы в максимально ранние сроки исключить атипичный ГУС и ГУС, связанный с пневмококковой инфекцией.
В отношении STEC-ГУС (типичного) рекомендуется адекватная симптоматическая терапия со своевременным подключением диализа при необходимости. Прогноз при этом в основном зависит от продолжительности анурического периода и сопутствующих повреждений ЦНС.
Атипичный ГУС чаще всего имеет в основе генные мутации, приводящие к дисфункции каскада комплемента с неконтролируемой активацией альтернативного пути. При общем неблагоприятном прогнозе этой склонной к рецидивированию формы перспективным является лечение экулизумабом, блокирующим терминальные компоненты каскада комплемента.
А.Н. Цыгин, Т.В. Вашурина, Т.В. Маргиева, П.В. Ананьин,
А.М. Мазо, А.А. Пушков, К.В. Савостьянов
ГЕМОЛИТИКО-УРЕМИЧЕСКИЙ СИНДРОМ (ГУС) является одной из частых причин острой почечной недостаточности (ОПН) у детей.
ТРИАДА СИМПТОМОВ:
- Гемолитическая анемия (снижение уровня гемоглобина и эритроцитов в крови, с наличием фрагментированных эритроцитов – шизоцитов)
- Тромбоцитопения – снижение уровня тромбоцитов в крови
- Острая почечная недостаточность
У большинства (90-95%) детей отмечается так называемый типичный или постдиарейный ГУС (Д+ГУС).
Другая форма ГУС, называемая атипичный (аГУС), встречается гораздо реже (5-10 % всех случаев) и является результатом аномалии (чаще генетической) белков, регулирующих процесс активации комплементов.
Д+ГУС ИЛИ ПОСТДИАРЕЙНЫЙ ГУС
Д+ГУС развивается вследствие кишечной инфекции вызванная Е coli (кишечная палочка) которая продуцируют токсин – шигатоксин (STEC).
тмечается в основном в возрасте до 3 лет и редко до 6 месяцев.
Симптомы
- диарея (разжиженный стул, понос) в течение нескольких дней (у 90-95%). Иногда бывает кровь в стуле. ЕСЛИ ВЫ ЗАМЕТИЛИ КРОВЬ В СТУЛЕ, НЕЗАМЕДЛИТЕЛЬНО ОБРАТИТЕСЬ К ВРАЧУ.
- рвота (у 30-60%) и боли в животе
Манифестация ГУС начинается в среднем через 6 (в среднем 2-14) дней. Бледность, общее недомогание, слабость, изменение поведения, небольшая желтушность, уменьшение количества мочи после (кровавой) диареи должны насторожить врача в отношении ГУС.
ЕСЛИ У ВАШЕГО РЕБЕНКА КИШЕЧНАЯ ИНФЕКЦИЯ И ЧЕРЕЗ НЕСКОЛЬКО ДНЕЙ ВЫ ЗАМЕТИЛИ ТЕМНУЮ МОЧУ (КРАСНУЮ) С УМЕНЬШЕНИЕМ ЕЕ КОЛИЧЕСТВА, ИЛИ ЗАМЕТИЛИ ПАСТОЗНЫЕ ВЕКИ ИЛИ ОТЕК ЛИЦА, БЛЕДНОСТЬ С ЖЕЛТУШНОСТЬЮ НЕ ЗАМЕДЛЕТЕЛЬНО ОБРАТИТЕСЬ К ВРАЧУ!
РАЗВЕРНУТАЯ КАРТИНА ГУС
- гемолитическая анемия (снижение гемоглобина, шизоциты)
- тромбоцитопения
- лейкоцитоз
ОПН с повышением уровня сывороточного креатинина и азота мочевины. Приблизительно половина пациентов имеют тяжелую олигурию (снижение количества мочи) или анурию (полное отсутствие мочи), 50-60% нуждаются в остром диализе. При наличии хоть какого-нибудь количества мочи постоянно определяются микро- или макрогематурия (кровь в моче) и протеинурия (белок в моче).
ВНЕПОЧЕЧНЫЕ ОСЛОЖНЕНИЯ
- поражение центральной нервной системы
- поражение желудочно-кишечного тракта (воспаление толстого кишечника – колит и др.)
- отек поджелудочной железы
- поражение печени
- сердечные осложнения
Для уточнения диагноза срочно должны быть проведены соответствующие лабораторные исследования и незамедлительно начать лечение.
ПРОГНОЗ
В большинстве случаев в течение менее чем 1-2х недель исчезают гемолитическая анемия и тромбоцитопения, нормализуется диурез. 10% детей в среднем в течение 4-х месяцев достигают терминальной ХПН. Смертность в основном в результате поражения ЦНС (центральной нервной системы).
СОВЕТЫ РОДИТЕЛЯМ
ПРЕДОТВРАЩЕНИЯ ИНФИЦИРОВАНИЯ STEC (кишечная палочка содержащий шигатоксин) и развития ГУС
- рубленая говядина должна быть хорошо прожарена до приобретения на разрезе серого цвета
- дети до 3 лет не должны употреблять непастеризованные продукты (молоко, сыр, фруктовые соки)
- до приготовления пищи, особенно после манипуляций с рубленой говядиной, необходимо мыть руки
- дети, которые прикасались к крупному рогатому скоту и другим животным, должны после этого умыться, а также перед едой.
- для предотвращения контаминации мяса кишечным содержимым необходим контроль убоя скота. Важен надлежащий надзор и уход за водоснабжения
- антибиотики? Многочисленные исследования показали, что антибиотикотерапия в период диареи увеличивает риск развития ГУС, возможно в связи с освобождением шигатоксина в результате лизиса бактерий. Тем не менее, этот риск пока не доказан.
Catad_tema Наследственные и врожденные болезни - статьи
Атипичный гемолитико-уремический синдром (аГУС). Клинические рекомендации.
Атипичный гемолитико-уремический синдром (аГУС)
МКБ 10: D59.3
Год утверждения (частота пересмотра): 2014 (пересмотр каждые 5 лет)
ID: КР550
Профессиональные ассоциации:
- Научное общество нефрологов России
- Российская ассоциация нефрологов
Утверждены
Согласованы
Ключевые слова
- Тромботическая микроангиопатия;
- Гемолитико-уремический синдром;
- Атипичный гемолитико-уремический синдром;
- Комплемент-опосредованная ТМА;
- Экулизумаб;
- Свежезамореженная плазма;
- Плазмообмен.
Список сокращений
АГ – артериальная гипертония
аГУС – атипичный гемолитико-уремический синдром
ГУС – гемолитико-уремический синдром
ЖКТ – желудочно-кишечный тракт
ЗПТ – заместительная почечная терапия
ЛДГ – лактатдегидрогеназа
МАГА – микроангиопатическая гемолитическая анемия
МАК – мембраноатакующий комплекс
НФГ – нефракционированный гепарин
ОПП – острое почечное повреждение
ПО – плазмообмен
ПЦР – полимеразная цепная реакция
СЗП – свежезамороженная плазма
ТМА – тромботическая микроангиопатия
ТПН – терминальная почечная недостаточность
ТТП – тромботическая тромбоцитопеническая пурпура
ЦНС – центральная нервная система
Термины и определения
- Атипичный гемолитико-уремический синдром (аГУС) – хроническое системное заболевание генетической природы, в основе которого лежит неконтролируемая активация альтернативного пути комплемента, ведущая к генерализованному тромбообразованию в сосудах микроциркуляторного русла (комплемент-опосредованная тромботическая микроангиопатия);
- Гаптоглобин – белок, связывающий свободный гемоглобин, который попадает в кровь при повреждении эритроцитов. Является чувствительным лабораторным маркером гемолиза.
- Мембраноатакующий комплекс (МАК) – терминальный комплекс комплемента, сформированный из С5b-9 компонентов;
- Мембранный кофакторный протеин (MCP) – интегральный трансмембранный белок, который экспрессируется на поверхности клеток, где связывает С3b и является дополнительным кофактором для CFI;
- Пенетрантность - частота проявления гена. Определяется по проценту особей в популяции из числа несущих ген, у которых он проявился. При полной пенетрантности доминантный или гомозиготно-рецессивный аллель проявляется у каждой особи, а при неполной пенетрантности – у части особей;
- Тромбомодулин (thrombomodulin, THBD) – эндотелиальный гликопротеин с антикоагулянтными, противовоспалительными и цитопротективными свойствами;
- Тромботическая микроангиопатия (ТМА)- клинико-морфологический синдром, характеризующий поражением сосудов микроциркуляторного русла. Морфологически ТМА проявляются в виде образования тромбов в микроциркуляторном русле, поражением эндотелия сосудов, клинически это проявляется в возникновении микроангиопатической гемолитической анемии, тромбоцитопении и признаках ишемии органов.
- Тромботическая тромбоцитопеническая пурпура (ТТП)- системная форма ТМА, в основе которой лежит тромбообразование в микроциркуляторном русле ряда органов, опосредованное сверхкрупными мультимерами фактора фон Виллебранда в условиях дефицита ADAMTS13
- Фактор комплемента H (CFH) - основной регуляторный фактор альтернативного пути активации комплемента, участвующий в расщеплении С3 конвертазы альтернативного пути и защищающий эндотелий от атаки комплемента;
- Фактор комплемента I (CFI) – сериновая протеаза, которая расщепляет С3конвертазу альтернативного пути активации комплемента;
- STEC-ГУС – ГУС, опосредованный инфекцией Escherichia coli, продуцирующей. шигатоксин (Shigatoxine; Stx) (Stx–продуцирующая E coli, STEC).
1. Краткая информация
1.1 Определение
Атипичный гемолитико-уремический синдром (аГУС) – хроническое системное заболевание генетической природы, в основе которого лежит неконтролируемая активация альтернативного пути комплемента, ведущая к генерализованному тромбообразованию в сосудах микроциркуляторного русла (комплемент-опосредованная тромботическая микроангиопатия).
Комментарии: Наряду с типичным ГУС и ТТП аГУС является классическим заболеванием из группы ТМА. В настоящее время ТМА рассматривают как клинико-морфологический синдром, характеризующий поражение сосудов микроциркуляторного русла.
Гистологически ТМА – это особый тип повреждения сосудов, представленный отеком эндотелиальных клеток с их отслойкой от базальной мембраны, расширением субэндотелиального пространства с накоплением в нем аморфного мембраноподобного материала и образованием тромбов, содержащих тромбоциты и фибрин, что приводит к окклюзии просвета сосуда, вызывая развитие ишемии органов и тканей.
Клинически ТМА проявляется тромбоцитопенией, развивающейся вследствие потребления тромбоцитов в процессах распространенного тромбообразования, микроангиопатической гемолитической Кумбс-негативной анемией (механический гемолиз), лихорадкой и поражением различных органов, главным образом, почек и центральной нервной системы (ЦНС).
В настоящее время достигнуты значительные успехи в понимании этиологии и особенно патогенеза ТМА, что позволило отказаться от старой номенклатуры её классических форм. Так, типичный ГУС, ранее называвшийся постдиарейным или Д+ГУС, следует называть STEC-ГУС (Shiga-Toxin продуцирующая Esherihia Coli), атипичный ГУС, ранее именовавшийся «неассоциированным с диареей» или Д-ГУС, получил название «комплемент-опосредованной ТМА».
1.2 Этиология и патогенез
Регуляция системы комплемента в норме
Активация системы комплемента осуществляется тремя основными путями: классическим, лектиновым и альтернативным. Общей точкой, на которой сходятся все три пути, является расщепление С3 компонента комплемента. Нарушения, лежащие в основе развития аГУС, касаются альтернативного пути активации.
В отличие от первых двух путей, активация которых начинается после связывания с иммунными комплексами или микроорганизмами, альтернативный путь находится в состоянии постоянной активации, исходный уровень которой низок (т.н. механизм «холостого хода»), что обеспечивается спонтанным гидролизом С3 компонента комплемента. Образующийся при этом фрагмент С3b может связываться как с патогенами, так и с собственными клетками организма. На чужеродной (например, бактериальной) поверхности С3b связывается с фактором В (CFB), в результате чего образуется С3-конвертаза (комплекс С3bВb). Последняя многократно усиливает расщепление С3 за счет формирования т.н. «петли амплификации». При присоединении к С3-конвертазе дополнительных фрагментов С3b образуется С5-конвертаза (С3bВb(С3b) – энзиматический комплекс, расщепляющий С5-компонент комплемента. При расщеплении С5 образуется С5b фрагмент, запускающий сборку мембраноатакующего комплекса С5b9 (МАК), который вызывает лизис бактериальных клеток.
Поверхность клеток хозяина в норме защищена от локальной амплификации и депозиции С3b. Эту защиту обеспечивает жесткий контроль со стороны ряда регуляторных факторов комплемента, представленных как плазменными, так и мембраносвязанными белками, фиксированными на поверхности эндотелиальных клеток. Основными плазменными протеинами, регулирующими альтернативный путь активации комплемента, служат факторы Н (CFH) и I (CFI), мембраносвязанными – мембранный кофакторный протеин (MCP) и тромбомодулин (THBD).
Фактор комплемента H (CFH) - основной регуляторный фактор альтернативного пути активации комплемента. Он блокирует образование С3-конвертазы и напрямую ускоряет ее распад. Кроме того, фактор Н является кофактором CFI в инактивации C3b, которая приводит к образованию неактивного фрагмента iC3b, неспособного связываться с фактором В для образования С3-конвертазы. Молекула CFH имеет две области связывания С3b. Первая локализуется в N-концевой части, где связывание С3b регулирует амплификацию альтернативного пути комплемента в плазме. Вторая область связывания находится в С-концевой части молекулы, в 19 и 20 экзонах, связывание с которыми нарушает способность С3b фиксироваться на поверхности эндотелия, что приводит к локальной инактивации альтернативного пути. Таким образом, CFH принадлежит ключевая роль в защите эндотелиальных клеток от активации комплемента. Кроме того, установлено важное значение CFH в регуляции функции тромбоцитов за счет блокирования активации комплемента на их поверхности, что, в свою очередь, приводит к уменьшению функциональной активности и способствует снижению риска тромбообразования.
Фактор комплемента I (CFI) – сериновая протеаза, которая расщепляет С3b, приводя к формированию неактивного iC3b в присутствии растворимых и/или мембраносвязанных кофакторов.
Мембранный кофакторный протеин (MCP) – интегральный трансмембранный белок, который экспрессируется на поверхности клеток, где связывает С3b и является дополнительным кофактором для CFI.
Тромбомодулин (th romb omod ulin, THBD ) – эндотелиальный гликопротеин с антикоагулянтными, противовоспалительными и цитопротективными свойствами, который служит также регуляторным белком системы комплемента, выполняя функции мембраносвязанного кофактора CFI. Связывает С3b, ускоряя его инактивацию CFI в присутствии CFH.
Таким образом, регуляция альтернативного пути комплемента осуществляется четырьмя белками – факторами H, I, MCP и THBD, взаимодействие которых приводит к преобразованию C3b в неактивную молекулу iC3b, блокируя тем самым ключевой механизм активации – образование все бо льших количеств С3-конвертазы с последующей безудержной продукцией МАК.
Система комплемента при аГУС
Атипичный ГУС – заболевание, основу которого составляет генетически обусловленный дефект регуляции альтернативного пути комплемента, результатом чего является его хроническая неконтролируемая активация. Предполагается, что имеющиеся у пациентов с аГУС мутации в генах, кодирующих регуляторные белки (CFH, CFI, MCP, THBD), приводят к нарушению защиты эндотелиальных клеток от активации системы комплемента вследствие дефицита или, чаще, функциональных нарушений этих протеинов. В результате этого на поверхности клеток эндотелия усиливается образование МАК, вызывающее их повреждение с обнажением субэндотелиального матрикса, трансформацией атромботического фенотипа в протромботический и последующим образованием тромбов. Дополнительный вклад в процесс тромбообразования у пациентов с мутациями фактора Н может вносить также активация комплемента на поверхности тромбоцитов, приводящая к усилению их функциональной активности.
Наряду с наиболее часто встречающимися мутациями белков-регуляторов, приводящих к нарушению их функции по контролю за активностью альтернативного пути комплемента (loss-of-function), описаны также мутации фактора В и С3 компонента комплемента, обеспечивающие значительное нарастание активности (gain-of-function) за счет стабилизации С3-конвертазы и её резистентности к инактивации, соответственно, что вызывает избыточную активацию системы комплемента. Преимущественное поражение почек при аГУС, по-видимому, обусловлено особенной чувствительностью фенестрированного гломерулярного эндотелия к повреждению, обусловленному нарушенной регуляцией комплемента.
Генетические аномалии при аГУС
У пациентов с аГУС наиболее часто (около 30% случаев) обнаруживают мутации гена CFH. К настоящему времени идентифицированы более 100 мутаций CFH у детей и взрослых пациентов с аГУС, причем не только наследственным, но и спорадическим. Большинство мутаций локализованы вС-концевых экзонах (19-20) молекулы CFH. Гомозиготные мутации CFH редки, чаще всего встречается носительство одной гетерозиготной мутации, однако возможно носительство двух и более гетерозиготных полиморфизмов фактора Н. Меньшее число мутаций (т.н. мутации 1типа) ассоциировано с количественным дефицитом CFH (низкий уровень CFH в плазме), однако в большинстве случаев мутации (особенно в С-терминальных экзонах) сопряжены с функциональным дефицитом фактора (мутации 2 типа). Носители этих мутаций имеют нормальную концентрацию CFH в плазме. Уровень С3 в плазме снижен у 30-50% пациентов с гетерозиготными мутациями CFH и более часто – при мутациях 1 типа. Пациенты с гомозиготными мутациями имеют наиболее низкие уровни как CFH, так и С3 в плазме.
Прямая связь концентраций CFH и С3 между собой отсутствует: уровень С3 может быть низким при нормальном уровне CFH и наоборот.
Около 10% больных аГУС (в большинстве своем – дети) имеют мутации в гене, кодирующем MCP.
Примерно у 10% пациентов выявляют мутации CFI. Описаны мутации гена фактора I, аналогичные таковым CFH, т.е. приводящие либо к нарушению синтеза, либо к нарушению функции этого фермента.
Мутации гена тромбомодулина отмечаются у 3-5% больных.
Кроме того, у небольшого числа пациентов описаны мутации фактора В (1-4% больных) и C3 компонента комплемента (2-10% больных), приводящие к их избыточной активации. Около 12% больных аГУС имеют мутации двух и более генов системы комплемента.
Анти- CFH-аутоантитела
Приблизительно у 6-10% пациентов с аГУС, преимущественно у детей, выявляют аутоантитела к фактору Н. Их действие направлено против С-терминального региона (экзоны 19-20) молекулы CFH, в связи с чем приводит к тем же последствиям, что и мутации фактора Н. Наличие подобных антител связано с дефицитом фактор Н-связанных белков 1 и 3 (CFHR1 и CFHR3), возникающим вследствие мутаций соответствующих генов. Установлено, что у 90% больных с анти-CFH-аутоантителами полностью отсутствуют CFHR1 и CFHR3, что обусловлено гомозиготной делецией в генах этих протеинов. Примерно у трети больных с аутоантителами к фактору Н выявлены также мутации различных факторов комплемента. Плазменная концентрация С3 снижена у 40-60% больных аГУС с анти-CFH-аутоантителами, причем в большей степени у тех, кто имеет более высокий их титр. Особенностью аГУС, обусловленного антителами к фактору Н, является частое рецидивирование заболевания.
Комментарии: Пенетрантность всех мутантных генов перечисленных факторов составляет около 50%. Следует подчеркнуть, что в семьях с идентифицированными мутациями генов комплемента только у некоторых носителей мутаций развивается аГУС. Даже при возникновении заболевания у нескольких членов семьи с одними и теми же мутациями его клинические проявления могут различаться. Значительный полиморфизм клинических проявлений наблюдается также не у родственных носителей тех же мутаций. Это дает основания предполагать наличие дополнительных факторов – генетических или средовых – которые могут оказать влияние на развитие или прогрессирование аГУС.
Таким образом, следует помнить, что:
- отсутствие семейной истории аГУС не исключает возможности генетической природы заболевания;
- практически невозможно предсказать риск возникновения аГУС у членов семьи больного, имеющих ту же мутацию, что и пробанд.
Генетически-фенотипические корреляции и прогноз аГУС
У пациентов с аГУС для постановки диагноза и принятия решения о тактике лечения идентификация мутаций факторов комплемента не требуется. Однако генетическое исследование необходимо для определения прогноза, особенно у больных, которым планируется трансплантация почки.
Комментарии: У взрослых пациентов с аГУС возраст дебюта заболевания не зависит от характера мутаций. Однако у детей последний определяет возраст начала заболевания. Так, у большинства детей с мутациями факторов Н и I, а также С3 и тромбомодулина болезнь дебютирует в возрасте до 5 лет. Дети, у которых выявлены антитела к фактору Н или мутации гена МСР, чаще заболевают в возрасте 8 лет и старше. Наличие мутаций в генах того или иного фактора комплемента непосредственно влияет на характер течения аГУС, общий и почечный прогноз, а также определяет прогноз у пациентов с трансплантированной почкой (Таблица 1). Таким образом, у пациентов с аГУС прогноз варьирует в зависимости от генотипа. Так, пациенты с мутациями CFH имеют худший прогноз, а больные с мутациями МСР – лучший. Смертность во время первого эпизода болезни составляет у детей с мутациями фактора Н 20-30%, а у взрослых – 4%. Терминальной почечной недостаточности (ТПН) в исходе острого эпизода среди выживших детей достигают 20-40%, среди взрослых – 48%. По сравнению с ними, не зафиксировано случаев смерти в момент острого эпизода болезни ни у одного больного с мутациями МСР независимо от возраста. ТПН развилась у 25% взрослых пациентов с этими мутациями, тогда как среди детей прогрессирования до ТПН не отмечено ни в одном случае. При естественном течении аГУС заболевание отличается неблагоприятным прогнозом независимо от того, в каких регуляторных белках и компонентах комплемента идентифицированы мутации, а также в случаях, когда выявить их не удалось.
Таблица 1. Прогноз при аГУС в зависимости от вида мутаций в генах комплемента
|
Риск смерти или ТПН в момент острого эпизода или через год от дебюта |
Риск рецидива |
Риск смерти или ТПН через 3-5 лет от начала болезни |
Риск рецидива после трансплан-тации почки |
|
|
3/3 без ТПН |
||||
|
1 больной |
||||
|
Анти- CFH-АТ |
Выше у б-х с высоким уровнем |
1.3 Эпидемиология
- аГУС – ультраредкое (орфанное) заболевание с распространенностью около 10% от распространенности STEC-ГУС, что составляет 2-7 случаев на 1000000;
- Заболевание может развиваться в любом возрасте, однако чаще поражает детей и молодых взрослых. Среди заболевших 60% составляют дети, 40% - взрослые;
- аГУС одинаково часто развивается у мужчин и женщин. При манифестации в более старшем возрасте болезнь несколько чаще поражает женщин.
1.4 Кодирование по МКБ 10
D59.3 – Гемолитико-уремический синдром
1.5 Классификация
аГУС принадлежит к группе первичных ТМА.
Классификация тромботических микроангиопатий (ТМА)
Тромботические микроангиопатии классифицируют на первичные и вторичные. Первичные ТМА включают в себя тромботическую тромбоцитопеническую пурпуру (ТТП), типичный ГУС и атипичный ГУС, этиология и патогенез которых установлены.
- ТТП, обусловленная аномалиями ADAMTS-13 (активность менее 5%);
Генетическими;
Приобретенными (аутоантитела, прием тиклопидина или клопидогреля);
- ГУС , индуцированный инфекцией:
- типичный ГУС = STEC-ГУС: шига (STEC)- и веротоксин (VTEC)- продуцирующими бактериями - энтерогеморагической Е.coli, штамм О 157:Н7 и другие штаммы, а также Shigella dysenteriae I типа, Streptococcus рneumoniae , продуцирующим нейраминидазу
- Атипичный ГУС , обусловленный генетическими нарушениями или изменениями иммунной системы, приводящими к патологии системы комплемента:
- - Мутациями генов регуляторных белков и компонентов комплемента CFH (фактор H), MCP (мембранный кофакторный протеин), CFI (фактор I), THBD (тромбомодулин), CFB (фактор B), и C3;
- - Антителами к CFH.
Наряду с первичными, существуют многочисленные вторичные ТМА, развитие которых связано с различными заболеваниями или состояниями. К ним относятся:
- Беременность и роды: преэклампсия-эклампсия, HELLP-синдром;
- Аутоиммунные заболевания: системная красная волчанка (СКВ), системная склеродермия, антифосфолипидный синдром (АФС);
- Злокачественные опухоли;
- Инфекции: ВИЧ, грипп H1N1;
- Другие заболевания: злокачественная артериальная гипертензия, гломерулопатии;
- Метилмалоновая ацидурия с гомоцистеинурией;
- Лекарственная терапия: хинин, интерферон, ингибиторы кальцинейрина (циклоспорин, такролимус), ингибиторы mTOR (сиролимус, эверолимус), противоопухолевые препараты (цисплатин, гемцитабин, митомицин, ингибиторы VEGFи тирозинкиназы – бевацизумаб, сунитиниб, сорафениб), оральные контрацептивы, валациклавир;
- Ионизирующее излучение;
- Трансплантация солидных органов и костного мозга. Комментарии: В ряде случаев возможно сочетание у пациента нескольких причин, ведущих к развитию ТМА. В частности, установлено, что 22% больных STEC-HUS имеют мутации генов системы комплемента, поэтому в подобных случаях STEC-инфекцию скорее следует рассматривать как триггерный механизм активации комплемента, приведший к развитию аГУС, чем как самостоятельное заболевание. Недавно было показано, что при ГУС, ассоциированном с беременностью, более чем у трех четвертей пациенток выявляют мутации регуляторных белков системы комплемента. Активация комплемента характерна для некоторых системных заболеваний, в первую очередь, СКВ и АФС, что позволяет рассматривать их как заболевания, коморбидные аГУС. Таким образом, следует считать генетические аномалии комплемента не причиной, а фактором, предрасполагающим к развитию ТМА.
Классификация аГУС
а ГУС подразделяют на:
- Семейный;
- Спорадический.
Комментарии: В структуре аГУС на долю семейного (диагностируемого по крайней мере у двух членов семьи) приходится, по разным данным, всего 10 - 20%, тогда как спорадический аГУС, при котором отсутствует семейный анамнез, встречается у 80-90% пациентов с этой патологией. Следует помнить, что отсутствие заболевания у родственников не исключает его наследственный характер.
2. Диагностика
Для развития аГУС необходимо взаимодействие генетических аномалий в системе комплемента с факторами внешней среды, которые играют роль триггеров, провоцирующих дополнительную активацию комплемента у предрасположенных лиц.Триггерные факторы идентифицированы в целом почти у половины пациентов с аГУС, при этом в педиатрической когорте больных частота их выявления достигает 80%. Наиболее частым триггером служат инфекции: в первую очередь, дыхательных путей и желудочно-кишечного тракта (ЖКТ). Диарея предшествует началу заболевания примерно в 23-30% случаев, причем в части из них выявляется STEC-инфекция. Среди пациентов со STEC-инфекцией, обследованных в связи с подозрением на аГУС из-за фульминантного течения болезни, наличия семейного анамнеза или рецидивов ТМА, генетические дефекты идентифицированы в 22% случаев. Инфекции верхних дыхательных путей как триггер аГУС зарегистрированы у 18% больных. Нередко заболеваниями, предшествующими развитию аГУС, являются грипп H1N1 и ветряная оспа. Важным фактором, способствующим развитию или рецидиву аГУС, служит беременность, которая предшествует заболеванию примерно у 7% больных, и трансплантация органов – у 5% пациентов.Клиническая картина аГУС характеризуется значительным полиморфизмом симптомов. Однако основными проявлениями болезни являются тромбоцитопения, микроангиопатическая гемолитическая анемия (МАГА) и острое почечное повреждение (ОПП), составляющие классическую триаду ТМА.
2.1 Жалобы и анамнез
Сбор анамнеза при подозрении на развитие аГУС подразумевает тщательный расспрос, направленный на выявление наследственной предрасположенности и возможных триггеров острой ТМА (перенесенные инфекционные заболевания, применение любых лекарственных препаратов, хирургические вмешательства, у женщин - акушерская патология), в том числе и системных, и онкологических заболеваний.
- Клиническая картина аГУС характеризуется значительным полиморфизмом симптомов. Жалобы неспецифичны и могут включать в себя: слабость, быструю утомляемость, повышение температуры тела, уменьшение количества мочи или отсутствие ее, изменение цвета мочи, одышку, головные боли, расстройства зрения;
- аГУС в большинстве случаев начинается внезапно. Болезнь может манифестировать неспецифическими симптомами – слабостью, утомляемостью, общим недомоганием, гриппоподобным синдромом. У взрослых пациентов нередко (в 20% случаев) имеет место стертое начало с медленным прогрессированием;
- В большинстве случаев поражение почек манифестирует острой почечной недостаточностью с наличием олиго/анурии или без нее;
- Артериальная гипертензия развивается у большинства пациентов независимо от возраста вследствие перегрузки объемом при наличии олиго/анурии и/или гиперренинемии вследствие ишемии ткани почек, обусловленной ТМА;
- Генерализованный характер ТМА при аГУС обусловливает развитие экстраренальных признаков болезни, связанных с поражением микроциркуляторного русла различных органов и систем, в том числе головного мозга, сердца, лёгких, ЖКТ. Внепочечные проявления заболевания наблюдаются у 20% пациентов, из которых почти две трети имеют больше одного экстраренального признака;
- У большинства пациентов имеется выраженный отечный синдром, основными проявлениями которого служат массивные периферические отеки вплоть до анасарки и выпот в полостях (гидроторакс, гидроперикард, асцит). Причиной отеков является резко повышенная сосудистая проницаемость, индуцированная С3а и С5а компонентами комплемента через освобождение больших количеств гистамина;
- Почти у половины больных развивается поражение ЦНС разной степени выраженности (сонливость, раздражительность, судороги, нарушения зрения, гемипарез или гемиплегия, ступор, кома). В ряде случаев возможно развитие отека головного мозга, обусловленное повышенной сосудистой проницаемостью;
- У 40% больных развивается ТМА миокарда, основным проявлением которой может быть дилатационная кардиомиопатия с признаками постепенно нарастающей или острой сердечной недостаточности. Небольшое число (около 3%) пациентов с интрамиокардиальной ТМА демонстрирует развитие острого инфаркт миокарда, который может стать причиной внезапной смерти;
- Легочная ТМА может стать причиной развития геморрагического альвеолита или острого респираторного дистресс-синдрома взрослых. Нарастающая вследствие этих причин дыхательная недостаточность в ряде случаев требует применения искусственной вентиляции легких. Достаточно часто у пациентов с аГУС развиваются двусторонние инфильтраты в легких, что затрудняет верификацию диагноза и требует дифференциальной диагностики с васкулитами и инфекционной патологией;
- Поражение ЖКТ встречается приблизительно у 30% больных аГУС. Наиболее часто наблюдается поражение кишечника, проявляющееся диареей, тошнотой и рвотой, хотя возможно развитие абдоминального болевого синдрома. Нередко отмечается развитие острого панкреатита с характерными клиническими признаками вплоть до панкреонекроза. Описано острое развитие сахарного диабета. Реже наблюдаются ишемические некрозы печени;
- Редким проявлением аГУС является поражение кожи с развитием обширных некротических очагов. Встречается также дигитальная ишемическая гангрена, приводящая к ампутации пальцев рук и ног;
- Примерно у 5% пациентов отмечается полиорганная недостаточность, связанная с диффузной ТМА с поражением ЦНС, ишемией миокарда, легочным кровотечением и дыхательной недостаточностью, панкреатитом, печеночным цитолитическим синдромом, желудочно-кишечным кровотечением.
2.2 Физикальное обследование
Общий осмотр подразумевает оценку общего физического состояния, роста (у детей) и массы тела, следов внутривенных инъекций. Необходимо обращать внимание на состояние подкожной жировой клетчатки, мышечной системы, на характерные для генетических нефропатий стигмы дисэмбриогенеза. Физикальное обследование должно включать:
- Осмотр кожных покровов для оценки наличия и степени выраженности отеков, изменения окраски (желтуха, эритема), высыпаний, экхимозов, геморрагий;
- Оценку сознания (возбуждение, судорожные припадки, заторможенность);
- Пальпацию лимфатических узлов, увеличение которых характерно для инфекционных заболеваний, болезней крови, онкологических заболеваний;
- Традиционное обследование всех органов и систем (аускультация легких и сердца, живота);
- Измерение АД.
- Пальпацию живота, зачастую позволяющую выявить увеличение печени, селезёнки, опухоль органов брюшной полости и забрюшинного пространства.
2.3 Лабораторная диагностика
Диагноз атипичного ГУС – это диагноз исключения. Он устанавливается на основании характерной клинической картины и должен быть подтвержден лабораторными данными, исключающими другие ТМА.
В связи с тем, что все ТМА независимо от их патогенеза имеют сходные клинико-лабораторные проявления и общую гистологическую картину, чрезвычайно важной представляется дифференциальная диагностика между основными формами первичной ТМА – ТТП, STEC-ГУС и аГУС. У взрослых пациентов с ТМА необходимо также исключить значительное число заболеваний и состояний, при которых возможно развитие вторичных ТМА, в первую очередь, связанных с беременностью и родами, системными заболеваниями (СКВ, АФС, склеродермия), злокачественными новообразованиями, ВИЧ-инфекцией, сепсисом, злокачественной артериальной гипертензией, лекарственной терапией, ДВС-синдромом. Таким образом, диагностика аГУС осуществляется в два этапа. На первом этапе необходимо установить наличие ТМА, на втором – провести дифференциальную диагностику между первичными и вторичными ТМА и первичных ТМА (ТТП, STEC-ГУС и аГУС) между собой.
- Рекомендовано критерями ТМА считать тромбоцитопению и микроангиопатическтй гемолиз (МАГА) в сочетании с признаками поражения почек и/или экстраренального поражения.
Комментарии: Тромбоцитопению констатируют при количестве тромбоцитов <150.000/мм3. Если число тромбоцитов превышает это значение, то об их потреблении можно судить по снижению количества тромбоцитов >25% от базального уровня (если он известен). В редких случаях возможно развитие МАГА без тромбоцитопении.
Наличие МАГА устанавливают на основании выявления у пациентов с анемией шизоцитоза (число шизоцитов в мазке периферической крови выше 0,1 %) и/или повышенного уровня (ЛДГ) и/или снижения гаптоглобина. При подозрении на ТМА необходимо определение всех трех указанных маркеров, поскольку в отсутствие изменений одного из них и не выполненных исследованиях двух других диагноз ТМА установить невозможно (ложноотрицательный результат!). Всем больным с ТМА необходимо также выполнять реакцию Кумбса для исключения иммунной природы гемолиза
У пациентов с МАГА и тромбоцитопенией наличие ОПП или других признаков поражения почек, изолированного или в сочетании с симптомами поражения ЦНС, сердца, ЖКТ, легких служит основанием для диагностики ТМА.
- В случае констатации ТМА диагноз аГУС рекомендовано верифицировать, только исключив STEC-ГУС и ТТП. Диагноз STEC-ГУС может быть отвергнут на основании исключения наличия шига-токсина в крови и стуле.
Комментарии: Скрининг на STEC-ГУС необходим всем больным с признаками поражения ЖКТ, особенно с диареей. Лабораторные исследования следует выполнять в первые сутки госпитализации больного в стационар до начала антибактериальной терапии. Для исключения STEC-ГУС показаны:
- посев кала для выявления культуры STEC (на среду Mac Conkey для E.coli O157:H);
- определение шига-токсина в кале или ректальном мазке методом полимеразной цепной реакции (ПЦР);
- определение шига-токсина в сыворотке крови;
- определение в сыворотке крови антител к липополисахариду наиболее распространенного в данном регионе серотипа E. Coli - O157:H7);
- Для исключения ТТП всем больным с ТМА рекомендовано определение активности ADAMTS-13.
Комментарии: Исследование активности ADAMTS-13 следует выполнять до начала плазмотерапии. Активность ADAMTS-13 в норме составляет 80-110%. Снижение её до 5% и менее свидетельствует в пользу диагноза ТТП. У больных с таким уровнем активности ADAMTS-13 показано определение в крови анти-ADAMTS-13-антител для верификации генетической или аутоиммунной формы ТТП, что определяет тактику лечения этого заболевания. У пациентов с аГУС активность ADAMTS-13 может быть снижена, однако её показатель всегда превышает 5%. Снижение активности ADAMTS-13, помимо аГУС, может наблюдаться при системных заболеваниях (катастрофический АФС, СКВ), сепсисе, ДВС-синдроме. В случаях невозможности немедленного исследования активности ADAMTS13 у пациента с ТМА и крайней тяжестью состояния, обусловливающей угрозу жизни или высокий риск тяжелых почечных и/или внепочечных осложнений, следует использовать правило, в соответствии с которым значения креатинина сыворотки >150-200мкмоль/л (1,7-2,3мг/дл) в сочетании с числом тромбоцитов >30 000/1мкл практически исключают диагноз ТТП.
Исключение STEC-ГУС и ТТП у пациента с не вызывающей сомнений ТМА позволяет диагностировать атипичный гемолитико-уремический синдром
- Всем пациентам с вновь выявленной ТМА рекомендовано исследовать кровь на содержание С3 и С4 компонентов комплемента.
Комментарии: Снижение содержания С3 при нормальном уровне в крови больных с клинико-лабораторными признаками ТМА С4 отмечается не более чем у 50% пациентов с аГУС. Нормальный показатель С3 не исключает диагноза аГУС, а выявленное снижение этого компонента комплемента может служить дополнительным аргументом в пользу этого диагноза.
- Больным с признаками ТМА, особенно детям и подросткам, рекомендовано выполнять исследование аутоантител к фактору Н (анти-FH-антитела).
Коментарий
Аутоантитела к фактору Н обнаруживают приблизительно у 10-15% больных аГУС, преимущественно у детей и подростков. Своевременное выявление анти-FH-антитела имеет важное значение для выбора тактики лечения
- Генетическое исследование не является необходимым для установления диагноза аГУС и не играет роли для решения вопроса о тактике лечения больного. Однако генетическое исследование рекомендовано для определения прогноза трансплантации почки, если она планируется, при семейных формах аГУС и рецидивах заболевания.
Комментарии: Отсутствие необходимости генетического исследования для диагностики аГУС основано на том, что мутации генов регуляторных белков альтернативного пути активации комплемента выявляются у больных с наследственным аГУС в 60-70% случаев, а при спорадической форме болезни – всего в 30%. Таким образом, отрицательный результат генетического скрининга у пациента с несомненной ТМА не исключает наличия аГУС. Выполнение генетического исследования занимает не менее 2х месяцев, а прогноз одинаков у больных как с идентифицированными, так и с неидентифицированными мутациями. Поэтому для диагностики аГУС и назначения лечения генетическое исследование значения не имеет. Однако после трансплантации почки риск рецидива аГУС, определяющего прогноз, зависит от вида мутаций (таблица 1). Именно поэтому при подготовке к трансплантации почки в план обследования пациента с аГУС необходимо включать генетическое исследование.
2.4 Инструментальная диагностика
- Рекомендовано проведение ЭКГ;ЭХО-КГ;Сцинтиграфия миокарда;УЗИ брюшной полости и почек;УЗДГ сосудов почек;МРТ головного мозга;Рентгенологическое исследование органов грудной полости;КТ легких и др.Инструментальная диагностика включает все методы, позволяющие уточнить характер поражения органов и систем, особенно при полиорганном повреждении:
2.5 Иная диагностика
- Рекомендовано биопсия почки для подтверждения диагноза ТМА в сомнительных и неясных ситуациях, однако она не является обязательной для диагностики аГУС.
Комментарии: Биопсия почки может помочь в верификации диагноза в следующих случаях: момнений в диагнозе аГУС; массивной протеинурии у больных с анемией и тромбоцитопенией;отсутствия эффекта от плазмотерапии; госпитализации в стационар в поздние сроки от дебюта ТМА;отсутствие полного лабораторного симптомокомплекса ТМА (чаще всего тромбоцитопении);подозрения на вторичные формы ТМА;подозрения на хроническую ТМА.
- Рекомендовано проводить дифференциальную диагностику аГУС с системными заболеваниями (СКВ, катастрофический АФС) и ВИЧ-инфекцией. Развитие симптомокомплекса ТМА во время беременности и в послеродовом периоде требует также исключения специфических акушерских причин данной патологии.
Комментарии: (Таблица 2) Взрослые больные и подростки с симтомокомплексом ТМА нуждаются в исключении системных заболеваний, в первую очередь, СКВ и АФС. Последний может развиться как в рамках СКВ (вторичный АФС), так и как самостоятельное заболевание (первичный АФС). Сочетание клинико-лабораторных проявлений ТМА с наличием антифосфолипидных антител безусловно свидетельствует в пользу диагноза «катастрофический АФС», независимо от того, имеются ли у пациента или отсутствуют клинические и иммунологические признаки СКВ. В связи с этим у больных с несомненным наличием ТМА следует обязательно определять серологические маркеры и СКВ, и АФС, поскольку выявленный у пациента спектр маркеров определяет терапевтическую тактику. Кроме системных заболеваний, необходимо исключать ВИЧ-инфекцию, так как среди пациентов с ВИЧ-инфекцией частота ТМА выше, чем в общей популяции, и возрастает с прогрессированием заболевания.
Развитие ТМА во время беременности и сразу после родов требует незамедлительной верификации диагноза, что определяет прогноз для матери и плода. «Акушерская ТМА» может быть представлена не только аГУС и ТТП (разграничение которых проводится теми же методами, что и вне беременности), но и специфическими гестационными видами патологии – преэклампсией и HELLP-синдромом, требующими незамедлительной верификации, так как от этого зависит выбор тактики лечения и прогноз.
Таблица 2. Дифференциальная диагностика аГУС у взрослых
|
Заболевание |
Дифференциально-диагностические признаки |
|
Типичный ГУС |
Положительный результат при бактериологическом исследовании кала: посев на среду для выявления STEC (Mac Conkey для O157:H7), определение в образцах фекалий ДНК энтерогеморрагических E.coli методом ПЦР; выявление в сыворотке антител к липополисахаридам наиболее распространенных в данном регионе серотипов E.coli. |
|
Наследственная или приобретенная ТТП |
Дефицит ADAMTS-13, антитела к ADAMTS-13 |
|
Беременность. Исключить HELLP-синдром преэклампсию, ТТП |
Тест на беременность, ферменты печени, срок гестации |
|
Аутоиммунные заболевания (системная красная волчанка, антифосфолипидный синдром) |
Анти-ДНК-антитела, антинуклеарные антитела, антикардиолипиновые антитела, анти-?2-ГП1-антитела, волчаночный антикоагулянт |
|
Положительные результаты иммунного блоттинга на ВИЧ-инфекцию |
|
|
ТМА на фоне злокачественных новообразований, химиотерапии (митомицин, блеомицин, цисплатин, ингибиторы VEGF), трансплантации, приема лекарственных препаратов (оральные контрацептивы, ингибиторы кальцийнейрина, тиклопидин, клопидогрель, интерферон, хинин) |
|
3. Лечение
Целями терапии аГУС, помимо обеспечения лучшей выживаемости больных, являются ингибиция неконтролируемой активации комплемента, купирование клинико-лабораторных проявлений ТМА, сохранение и улучшение функции пораженных органов (в том числе, предотвращение развития ТПН, избавление от потребности в диализных методах лечения, недопущение поражения других внутренних органов, кроме почек), улучшение качества жизни пациентов.
- Рекомендована госпитализхация в многопрофильные стационары с хорошо оснащенным отделением реанимации и интенсивной терапии больных с признаками ТМА.
Комментарии: Госпитализация в подобные стационары обусловлена необходимостью применения диализных методов лечения (гемодиализ, продленная веновенозная гемодиафильтрация), искусственной вентиляции легких (ИВЛ) и плазмотерапии (ПТ).
3.1 Консервативное лечение
Консервативное лечение аГУС включает:
- Плазмотерапию;
- Препарат группы комплемент-ингибирующих антител (Экулизумаб**);
- Препараты эритропоэтина**;
- Трансфузии эритроцитарной массы;
- Антигипертензивные препараты;
- Антибактериальную и противовирусная терапия (по показаниям).
- Рекомендовано всем взрослым больным с впервые выявленной ТМА или рецидивом аГУС, диагноз которого был установлен ранее, назначается плазмотерапия (ПТ). Плазмотерапия может проводиться в режимах инфузий свежезамороженной плазмы (инфузия плазмы) или плазмообмена (ПО), причем режим плазмообмена более предпочтителен.
Комментарии: Хотя проспективные клинические исследования по лечению аГУС свежезамороженная плазма (СЗП) отсутствуют, ПТ в течение нескольких десятилетий эмпирически используется как предпочтительный метод лечения этого заболевания, поскольку накопленный за этот срок опыт свидетельствует об уменьшении летальности пациентов с аГУС под действием плазмотерапии. При этом частота восстановления гематологических показателей и функции почек не превышает 50%. При инфузии плазмы пациенту вводится донорская СЗП, содержащая функционально активные регуляторные белки системы комплемента, что устраняет дефицит собственных естественных регуляторов – факторов H и I, а также прекращает тромбообразование в сосудах микроцируляторного русла за счет введения естественных компонентов плазмы, обладающих протеолитической активностью в отношении сверхкрупных мультимеров ф.В, антикоагулянтов и компонентов системы фибринолиза. При ПО, кроме этого, происходит удаление измененных эндогенных циркулирующих ингибиторов комплемента и, возможно циркулирующих антител к фактору Н. Режим ПО предпочтительнее ИП, поскольку позволяет вводить бо льшие объемы СЗП без развития гиперволемии и гипергидратации, что особенно важно у пациентов с олигурией, поражением ЦНС и сердца.
- Рекомендовано инфузии СЗП необходимо проводить в объеме 30-40 мл/кг в 1-й день, 10-20 мл/кг в последующие дни.
- Рекомендовано в начале терапии следует провести 5 сеансов ПО ежедневно с объемом эксфузии 40мл/кг/с, при необходимости 60-75 мл/кг/с с замещением адекватным объемом СЗП (1-1,5 расчетного объема плазмы). Затем в последующие 2 недели необходимо проводить по 5 сеансов ПО в том же режиме. Далее сеансы проводят через день (3 сеанса в неделю) еще в течение 2х недель.
Комментарии: При лечении аГУС возможно сочетание режимов инфузии плазмы и ПО.Продолжительность лечения СЗП не определена. Решение о продолжении ПТ следует принимать в зависимости от его эффективности. Сеансы ПО следует продолжить до нормализации числа тромбоцитов, прекращения гемолиза и улучшения функции почек. В связи с этим терапию СЗП необходимо контролировать ежедневным определением количества тромбоцитов и уровня ЛДГ.
Критериями эффективности ПТ служат:
исчезновение тромбоцитопении;
прекращение гемолиза, о чем свидетельствует нормализация ЛДГ;
Стойкая нормализация уровня тромбоцитов и прекращение гемолиза в течение не менее 2-3х дней является показанием к прекращению ПТ;
Следует помнить, что в ряде случаев нормализация гематологических показателей может не сопровождаться улучшением функции почек. Это дает основания для изменения тактики лечения.
Пациентам с аГУС без выраженной тромбоцитопении (число тромбоцитов менее 20000/мкл), не имеющим тяжелых осложнений (кровотечения), трансфузии тромбоконцентрата противопоказаны.
Трансфузии тромбоцитов могут усилить проявления ТМА у пациентов с аГУС, поскольку провоцируют новые эпизоды микротромбообразования с дальнейшим потреблением тромбоцитов.
- Рекомендовано при отсутствии выраженной тромбоцитопении и кровоточивости у больных аГУС ПТ следует сочетать с назначением гепаринотерапии.Во время острого эпизода ТМА обычный (нефракционированный) гепарин (НФГ) следует назначать внутривенно капельно с дозированной скоростью введения (через инфузомат). При таком режиме введения доза НФГ составляет 250-1000ЕД/час. Возможно также введение малых доз НФГ (2500-5000ЕД) непосредственно в емкость с СЗП перед инфузией.
Коментарии: Сочетание НФГ с ПТ позволяет усилить антитромботический эффект СЗП и способствует более быстрому купированию тромбоцитопении.
Пациентам с аГУС, имеющим выраженную МАГА (Нв менее 75г/л), необходима коррекция анемии.
С этой целью следует применять трансфузии эритроцитной массы и/или препараты эритропоэтина (См. рекомендации по лечению анемии у нефрологических больных).
Наличие у больных аГУС артериальной гипертензии требует назначения антигипертензивных препаратов
- Рекомендовано у пациентов с аГУС выявлять возможные триггеры заболевания и назначить лечение, направленное на их подавление.
Комментарии: Особое значение имеет своевременная диагностика инфекций, наиболее часто предшествующих аГУС, и следующая за ней адекватная антибактериальная терапия.
- Ркомендовано назначать Экулизумаб – препарат группы комплемент-ингибирующих антител взрослым пациентам с аГУС в случаях неэффективности плазмотерапии, плазмозависимости, развития нежелательных явлений в процессе ПТ, рецидивирующего течения заболевания или семейном его характере.
Комментарии: Экулизумаб – рекомбинантное гуманизированное моноклональное антитело класса Ig G к С5 компоненту комплемента. Экулизумаб блокирует расщепление С5 на С5а и С5b, что препятствует образованию мембрано-атакующего комплекса С5b-9 и подавляет провоспалительное, протромботическое и литическое действия комплемента, предотвращая повреждение эндотелия и прекращая процессы микроциркуляторного тромбообразования. Применение Экулизумаба приводит к обратному развитию ТМА и/или предупреждает прогрессирование поражения почек. Блокируя терминальный комплекс комплемента, Экулизумаб сохраняет интактным проксимальное звено каскада комплемента, что крайне важно для опсонизации микроорганизмов и клиренса иммунных комплексов.
Режим терапии экулизумабом:
- Рекомендовано за 2 недели до начала лечения Экулизумабом больного аГУС необходимо вакцинировать против Neisseria meningitidis, поскольку на фоне применения препарата возрастает риск развития менингита. Вакцинацию производят конъюгированной тетравакциной против серотипов возбудителя A,C,Y и W135.